5
Mutations
Professor A. Cuschieri
Department of Anatomy
University of Malta
OBJECTIVES:
By
the end of this session the student should be able to:
·
List the different types of mutation
·
Explain how point mutations can cause severe diseases
·
Give examples of deletions, duplications, and
insertions in genes
·
Define trinucleotide repeat expansions and how they
cause neurological diseases
Mutations
Mutations are permanent
changes in the sequences of genomic DNA. The mutations that are of main
interest to medical genetics are the ones that have deleterious effects on the
phenotype and cause disease conditions.
Recent advances in medical genetics led to the discovery of an
increasing number of mutations that are responsible for genetic diseases. Most mutations are inherited from one
generation to the next but a few arise de novo as new mutations. They may then be passed on to future
generations, following the rules of monogenic inheritance. Most new mutations
are spontaneous, occurring without any obvious cause but they occur
regularly. However, particular
mutagenic agents, which may be chemical, viral or radiation, can also cause
mutations. It is possible that undetected environmental agents cause the
so-called spontaneous mutations.
Mutations are the
source of genetic variation. Several
mutations do not exert any negative phenotypic effects and are labeled as
normal variants, while some mutations may even be beneficial. While most research in medical genetics has
been directed towards the identification of disease-causing mutations, such as
those that cause mental retardation, there is increasing interest in mutations
that cause unusually high intelligence in gifted individuals. These may be more difficult to detect.
There are several
different types of mutation that range from those affecting only one base pair
to those involving whole chromosomes or chromosome segments. The main types of mutations may be
classified under the following headings:
a)
Point Mutations
b) Deletions
c) Duplications
d) Insertions
e) Frame shift mutations
f) Trinucleotide repeat expansions
g) Chromosome abnormalities
Point mutations
A point mutation or single base pair
substitution is a change in one nucleotide.
A point mutation that
occurs in an exon of a coding gene results in an alteration of the transcribed
protein. If a point mutation occurs
within an intron of a coding gene, it does not cause any change in the coded
message, but may result in a defect in RNA splicing and in this way could also
affect the resulting protein. A point mutation may affect other non-coding
regions, such as a promoter sequence, resulting in failure of transcription of
the protein. It may also affect regulatory sequences, resulting in decreased
translation and decreased gene product.
A point mutation may also occur at random in non-coding DNA regions.
Such mutations are very common and are usually termed single
nucleotide polymorphisms (SNPs).
While they do not have any effect on the phenotype SNPs are nevertheless
very important, as they are useful genetic markers for identification of
individuals and in the determination of paternity.
The following are some
examples of point mutations occurring within exons of genes.
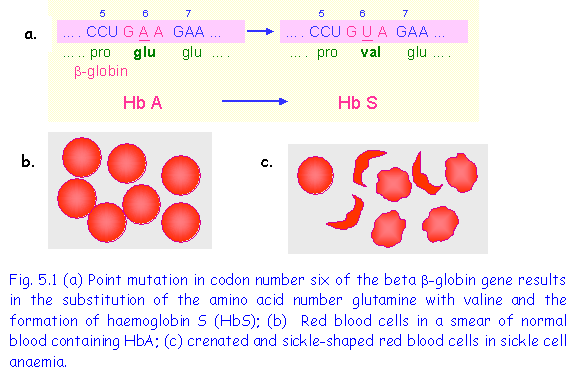
1. Point mutation causing Sickle Cell Anaemia
Sickle cell anaemia is the result of a point mutation in codon 6 of the b-globin gene resulting in the substitution of amino acid glutamic acid by valine (Fig 5.). b-globin
is a major component of adult haemoglobin (HbA). The single amino-acid substitution results in a type of haemoglobin termed HbS, which has different properties from the normal HbA. Under conditions of low oxygen tension, such as following exercise or in an atmosphere containing a low oxygen level, the following changes occur:
(1)The haemoglobin agglutinates to form insoluble rod-shaped polymers;
(2)Red blood cells become distorted and sickle-shaped (Fig 5.1);
(3)The sickle-shaped cells rupture easily causing haemolytic anaemia;
(4)The sickle shaped cells tend to block capillaries interfering with the blood flow to various organs.
2. Point Mutation In The b-Globin Gene Resulting In b Thalassaemia
(Fig.5.2)
In this example the substitution of C by U resulted in the formation of
a stop signal UAG in place of glutamate in codon number 39, and a shortened
globin chain contining only 39 instead of the normal 146 amino acids in the b-globin protein chain. This protein is functionally useless and is
equivalent to absence of b-globin gives clinical symptoms of b thalassaemia,

3. Point Mutation In The alpha-Globin Gene Results In Elongated a-Globin Chain (Fig 5.3).
This example contrasts with the previous one because the stop codon UAA at position 142 in the alpha (a-) globin gene was substituted by the codon for glutamine. Translation of the protein thus continued until a stop codon was encountered at codon 173. The a-globin was considerably elongated, resulting in a variant of haemoglobin termed haemoglobin Wayne.

Deletions
Deletions involve removal of one or more base pairs. They vary greatly in siz from deletion of a single base to deletion of a whole gene. The clinical effects often depend on the size and location of the deleted part of the gene. The following are some examples of deletions:
1. Deletions involving the dystrophin gene
Dystrophin is a protein that is an important component of skeletal muscle. The dystrophin gene is located on the long arm of the X chromosome. It is a very large gene spanning 2.5 million bp of genomic DNA and consists of 79 exons coding for a protein of approximately 3600 amino acids (11kb)
.
Deletion of the whole or most of the dystrophin gene (Fig 5.4-b) results
in Duchenne muscular dystrophy. This is
a severe X-linked recessive
disorder that affects boys and is transmitted by carrier females. In affected
boys there is almost complete lack of dystrophin, muscle weakness beginning in
childhood and increasing progressively in severity so that the individual is
wheel-chair bound at the age of about 15 years. Death usually ensues in the
early twenties due to respiratory muscle involvement.
Deletions involving a
small non-critical part of the gene result in altered dystrophin that causes
the clinical condition of Becker muscular dystrophy in which muscle weakness
begins in adolescence and is very slowly progressive, and affected individuals
may lead an almost normal life.
It is evident that,
although Duchenne and Becker muscular dystrophy are both caused by a deletion
in the same gene, a female carrier of Becker muscular dystrophy be reassured
that she cannot have a son affect with the more severe Duchenne muscular
dystrophy.

2. Deletion of one codon causing
Cystic Fibrosis.
Cystic fibrosis is caused by a mutation in the Cystic Fibrosis Trans-membrane Regulator (CFTR) gene. This gene
Produces a
trans-membrane protein that regulates the flow of chloride ions into the
cells. The most common mutation is
termed the ∆508 mutation, which is a deletion of a single codon
at position number 508 in exon 10 of the CFTR gene.
Homozygotes for a ∆508 mutation have cystic fibrosis disease, which is characterized by the secretion of very thick, mucus causing obstruction of the bronchi and predisposing to pulmonary infections, pancreatic duct obstruction, intestinal and liver problems and excess salt loss in sweat. However, different deletions in the CFTR gene may affect the resulting clinical manifestations.
Some deletions may
cause mild forms of cystic fibrosis.
One type of mutation does not cause any symptoms of CF but cause
congenital bilateral absence of the vas deferens resulting in male infertility.
3. Deletion of 6 codons in the b-globin gene resulting in a variant haemoglobin
(Fig. 5.5).
In this example codons 92 to 97 of the b-globin gene are deleted. This results in a shortened b-globin protein that produces a haemoglobin variant termed Haemoglobin Gun Hill. In homozygotes it produces mild clinical; symptoms.

Insertion mutations
An example of an insertion is found in
haemophilia A, an X-linked recessive disorder in which blood clotting does not
occur due to deficiency of clotting factor VIII. In most cases the mutation is the result of insertion of a large segment, consisting of
about 3800 bp, in the coding region of
the factor VIII (Fig 5.6). This results
in total inactivation of the protein.

Frame shift mutations
Frame shift mutations involve a deletion or insertion of one or two base pairs within a coding sequence of a gene. As the coding message is read in triplets codons and deletions the reading frame of mRNA is altered resulting in a non-sense sequence of amino acids. An example occurs in the b-globin gene in which one nucleotide of codon 39 is deleted (Fig.5.7). The following reading frame is completely altered and continues until a stop codon is encountered. Some cases of Duchenne muscular dystrophy are caused by frame shift mutations in the dystrophin gene.
The part of the dystrophin molecule distal to the mutation is defective and non-functional.

Insertions
Insertion of a
sequence of bases into a coding sequence of a gene may occur. Sometimes a whole
gene sequence may be duplicated. For
example, in the condition known as hereditary motor and sensory neuropathy type
I, a DNA segment at locus 17p11 is duplicated.
Insertion of a small number of nucleotides that are not multiples of
three will result in frame shift mutation.
An example of this occurs in Tay Sach's disease in which insertion of 4 base pairs in exon 11 of the
hexoseaminidase gene, results in a
frame shift mutation distal to the insertion (Fig 5.8).

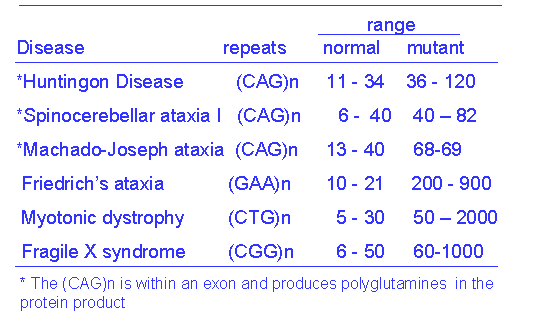
Trinucleotide repeat expansions
Trinucleotides are
triplets of nucleotides that are repeated in tandem many times over. The number
of repeats varies in different individuals.
An example of trinucleotide repeats is
- - - CAG CAG CAG CAG CAG - - - -, and is designated as (CAG)n where n
is the number of repeats in the particular individual. The number of repeats is usually stable and
the same number of repeats is transmitted from generation to generation. Trinucleotide repeats are widespread in the
genome, and may occur in exons, introns, promoter sequences or non-coding
regions. They are perfectly normal and
occur in all individuals. A mutation
arises when the repeats become unstable and undergo expansion, namely an
increase in the number of repeats as they are transmitted from one generation
to the next. When the number of repeats
exceeds a certain limit, clinical symptoms occur. An example is the huntingtin
gene, which, when mutated, causes Huntington's disease. Here the normal range of (CAG)n is 11 to 34,
and symptoms of Huntington's disease appear in individuals in whom the number of
repeats is greater than 37. The
intermediate range of 35 to 37 is a gray zone of uncertain significance.
The (CAG)n repeats in
the huntingtin gene occur within the exon and are therefore translated as
repeats of the amino acid glutamine (polyglutamine) within the huntingtin
protein. An excess of repeats causes
the protein to form aggregates that are deposited within the neurons causing
neuronal degeneration. There are
several other neurological disorders that are the result of trinucleotide
repeat expansions (Table 1). Some of them have the repeats in the exon and
express amino acid repeats within the protein.
Others affect the promoter sequence or introns and interfere with the
transcription of the gene.
Beta Thalassaemia
Beta thalassaemia is a genetic disorder in which there is lack of
beta globin.
It may be the result of:
i) deletion of the whole gene so that beta globin cannot not produced (designated bo )
(ii) deletion of the promoter region so that transcription cannot occur (designated bo )
iii) deletion of a large part of the gene or
iv) frame shift mutation resulting in a grossly abnormal or non-functional protein(designated b+ ).
Clinical Features of Beta-Thalassaemia
•haemoglobin A (a2 b2) cannot be produced
•Hb F (a2 g2) is produced even in adults
•Hb A2 (a2 d2) formation is increased
•erythrocytes are microcytic (small) due to lack of normal haemoglobin
•erythrocytes rupture easily causing severe haemolytic anaemia, requiring repeated blood transfusions
•the bone marrow expands trying to compensate by increasing haemopoiesis.
•the bones of the face and skull are
thickened causing a characteristic facial appearance
•the spleen and liver enlarge because haemopoietic tissue forms in them
•excess iron accumulates in the blood and is deposited in the heart, liver, pancreas and other organs (this is because of repeated transfusions while no blood is actually lost from the body)
•children have delayed growth and development and are prone to repeated infections
Carriers of Beta -Thalassaemia (heterozygotes)
Individuals are generally normal but have:
• haemoglobin at the lower limit of normal
• red blood cell count at lower limit of normal
• red blood cells are slightly microcytic
• HbA2 and Hb F may be slightly increased.
Although there is only one normal b globin gene, enough b globin is produced to synthesize adequate amounts of Hb A.
There are two pairs of alpha-globin genes.
Various situations arise depending on the
number of deleted genes
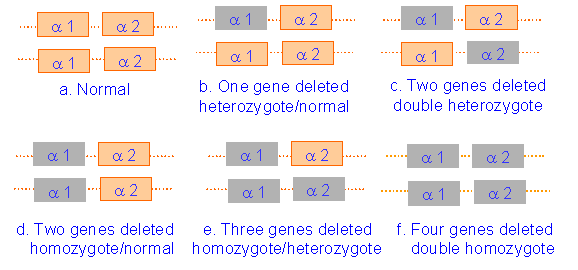
Features of Alpha Thalassaemia
In the extreme case when four alpha-globin
genes are deleted, the clinical features are:
• Very rare, and extremely severe disorder
• HbF, HbA or HbA2 cannot be produced (because all require alpha globin)
• An unusual haemoglobin termed beta-4 is formed
• There is very severe anaemia
• Baby is born with hydrops foetalis, which consists of
- massive oedema
- greatly enlarged liver and spleen
- always fatal; many babies are stillborn