Lysosomes
Professor Alfred Cuschieri
Department of Anatomy, University of Malta
Objectives
![]() Identify lysosomes in electron micrographs
Identify lysosomes in electron micrographs
![]() State the characteristics and functions of
lysosomes
State the characteristics and functions of
lysosomes
![]() Explain how lysosomes are formed
Explain how lysosomes are formed
![]() Name the properties of lysosomal membranes
Name the properties of lysosomal membranes
![]() Explain the mechanisms and importance of
phagocytosis
Explain the mechanisms and importance of
phagocytosis
![]() Give examples of phagocytic cells
Give examples of phagocytic cells
![]() Explain the importance of receptor-mediated
endocytosis
Explain the importance of receptor-mediated
endocytosis
![]() Describe how lysosomal enzyme deficiency
cause serious disease
Describe how lysosomal enzyme deficiency
cause serious disease
![]() Identify peroxisomes and state their
characteristic features
Identify peroxisomes and state their
characteristic features
![]() Explain the importance of peroxidase
generation and degradation in lysosomes
Explain the importance of peroxidase
generation and degradation in lysosomes
![]() Name some diseases caused by deficient
functions of lysosomes
Name some diseases caused by deficient
functions of lysosomes
![]() Distinguish between melanosomes and melanin
synthesis
Distinguish between melanosomes and melanin
synthesis
![]() Distinguish between melanosomes and
melanocytes
Distinguish between melanosomes and
melanocytes
Lysosomes
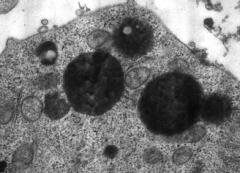
![]() are membrane bound organelles
are membrane bound organelles
![]() contain hydrolytic enzymes
contain hydrolytic enzymes
![]() have acidic contents (pH 4.5-5.5)
have acidic contents (pH 4.5-5.5)
![]() have electron-dense heterogeneous contents
have electron-dense heterogeneous contents
![]() digest ingested material and aged or damaged
organelles
digest ingested material and aged or damaged
organelles
Lysosomal enzymes are hydrolases that catalyse the reaction: AB + H2O à AH + BOH
Over
60 lysosomal enzymes are known.
There is
a hydrolyse for each type of biological molecule. These include:
Peptidases
– hydrolyse proteins
DNAases – hydrolyse DNA
RNAases
– hydrolyse RNA
Lipases – hydrolyse lipids
Phosphatases
– hydrolyse
phosphates
Glucosidases
– hydrolyse
glycogen
Carboxylases
- hydrolyse
carboxyl groups
Sulphatases
– hydrolyse
sulphates
Esterases
– hydrolyse
esters
Acid phosphatase and b-glucuronidase have been used as histochemical markers for lysosomes.
The lysosome membrane has three special properties:
1. an ATP driven proton [H+] pump to maintain a low pH (4.5-5.5) in the lysosomal compartment.
The
proton pump:
![]() Has a
‘lollipop’ structure similar to the F1 and Fo of mitochondria, and consisting of:
Has a
‘lollipop’ structure similar to the F1 and Fo of mitochondria, and consisting of:
![]() a head
containing 6 polypeptide units
a head
containing 6 polypeptide units
![]() a stalk
contains 5 polypeptide units
a stalk
contains 5 polypeptide units
![]() Is inhibited by n-ethylmaleimide
Is inhibited by n-ethylmaleimide
2. a
glycoprotein coat, rich in carbohydrates, on
its inner surface to protect it against hydrolysis by its own enzymes
3. transporter channels that transport
break-down products such as amino acids, glucose, nucleotides and other small molecules out of the lysosome. These
molecules may move out
a. by facilitated diffusion ;
b. by active transport;
c. by co-transport using the proton (H+) gradient to
provide the energy for transport.
The Formation of Lysosomes
a. The hydrolytic enzymes are formed in the RER
b. Enzymes have a terminal mannose–6-phosphate group as a marker to be packaged in lysosomes
c. They are packaged at the trans face of the Gogi complex (termed the Trans Golgi Network -TGN)
d. The newly formed vesicles that bud off are termed early endosomes or primary lysosomes. They are small and have homogeneous, electron-dense contents.
e. They early endosomes (primary lysosomes) may fuse with phagocytic or other vesicle to form late endosomes or secondary lysosomes.
Functions of
Lysosomes
Lysosomes may be involved in the following pathways:
1. phagocytosis
2. autophagocytosis
3. Formation of endosomes
4. Receptor-mediated endocytosis
1
Phagocytosis
Phagocytosis is the process by which the cell engulfs particulate matter (>0.5 mm) from the extra cellular space, and digests it by lysosomal action. Three main cell types are capable of performing this function:
a. Macrophages
i. are widely distributed in the connective tissue around the body
ii. phagocytose and digest all particulate matter in the extracellular space, including micro-organisms, foreign particles and damaged cells.
iii. are derived from monocytes, circulating in the peripheral blood.
iv. may have different names in different tissues such as Kupffer cells in the liver, osteoclasts in bone, and microglia in the central nervous system.
b. Neutrophils in the peripheral blood, which phagocytose and digest microorganisms.
c. Eosinophils in the peripheral blood, which phagocytose and digest antigen-antibody complexes.
Macrophages have a gylcocalyx coat, rich in glycosaminoglycans, on their outer surface, which causes particulate matter to adhere to them. Neutrophils and eosinophils have specific receptors to recognise specific particles to be engulfed. (The specific molecules that bind to the receptors are called ligands).
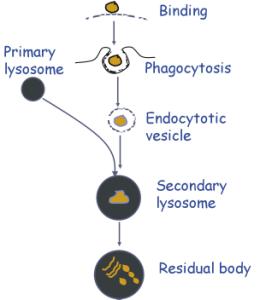 Phagocytosis involves:
Phagocytosis involves:
a. Adhesion of the particle to the glycocalyx or specific receptor on the plasma membrane
b. Extrusion of pseudopodia to surround the particle. This is mediated by actin filaments.
c. Formation of a phagocytic vacuole containing the engulfed particle
d. Fusion with a primary lysosome
Autophagocytosis
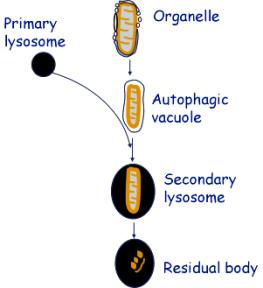 This is the process in which old or damaged organelles are broken
down. It occurs in practically all cells as a recycling system. It is most
marked in cells that are not replaced, such as neurons.
This is the process in which old or damaged organelles are broken
down. It occurs in practically all cells as a recycling system. It is most
marked in cells that are not replaced, such as neurons.
Autophagocytosis involves:
a. The organelle is surrounded by vesicles, which coalesce
b. The coalesced vesicles form a membrane surrounding the organelle. This is called an autophagic vacuole.
c. The autophagic vacuole fuses with one or more primary lysosomes to form secondary lysosomes.
d. Residual bodies are lysosomes with partially undigested material
Endocytosis and exocytosis
Endocytosis is the uptake of extra cellular fluid by infolding of the plasma membrane, and formation of a vesicle containing the extra cellular material. This process was formerly called pinocytosis.
Exocytosis is the reverse of pinocytosis, i.e. the extrusion of fluid contained in vesicles into the extra cellular space.
Endocytosis and exocytosis are important mostly for membrane flow. For example, exocytosis replaces the plasma membrane removed by phagocytosis. Similarly, exocytosis of secretion vesicles must be balanced by endocytosis.
Receptor-mediated endocytosis
![]() Receptor-mediated
endocytosis is the process whereby cells that have a specific receptor take up
specific macromolecules.
Receptor-mediated
endocytosis is the process whereby cells that have a specific receptor take up
specific macromolecules.
![]() This process is
used for the uptake of hormones, growth factors, antibodies, lipoproteins etc
This process is
used for the uptake of hormones, growth factors, antibodies, lipoproteins etc
![]() The receptors are
integral membrane proteins
The receptors are
integral membrane proteins
![]() The molecule that
binds to the receptor is termed the ligand.
The molecule that
binds to the receptor is termed the ligand.
The process of receptor-mediated endocytosis involves the following steps:
1
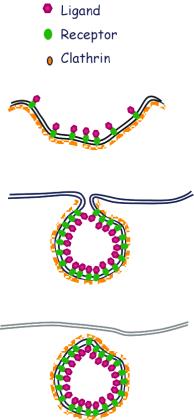 Binding of the ligand to the receptor
Binding of the ligand to the receptor
2 Lateral diffusion of the ligand-receptor complex
3 Accumulation of clathrin, adaptor protein and dynamin on the cytoplasmic surface of the plasma membrane at a particular site
4 Formation of a pit, and accumulation of the ligand receptor complex at the site of clathrin acculmulation
5 Deepening of the pit and formation of clathrin-coated vesicles containing the ligand-receptor complex
6 The vesicles lose their clathrin coat
7 The vesicles fuse with a primary lysosome (early endosome) and the ligand is cleaved from the receptor
Familial hypercholesterolaemia is a condition in which there is defective binding of low density lipoprotein (LDL) to its receptor. Receptor-mediated endocytosis of the LDL does not occur, and it accumulates as high levels in the blood.
The process of receptor-mediated
endocytosis was first described through the study of LDL uptake by cultured
fibroblasts.
Three molecules are involved in forming the coat on the cytoplasmic face of coated vesicles:
1.
Clathrin
§ A molecule that has a triskelion (three-pronged) structure.
§ Under appropriate conditions it forms a hexagonal (geodesic) latticework on the cytoplasmic surface of the plasma membrane,
§ It causes the membrane to invaginate and form a coated pit and vesiclevesicle
2.
Adaptor protein
§ Binds to the cytoplasmic end of the transmembrane receptor
§ Mediates the attachment of clathrin to the plasma membrane
§ Regulates clathrin assembly and disassembly
§ Is itself regulated by phosphorylation and dephosphorylation
3.
Dynamin
§ Incorporates a GTPase that provides the energy for formation of the coated pit and vesicle
§ Undergoes a configuration change that brings about closure of the vesicle
Gangliosidosis
is an example of a Lysosomal Storage Disease
Gangliosidosis:
![]() Is caused by deficiency of the enzymes b-galactosidase.
Is caused by deficiency of the enzymes b-galactosidase.
![]() Is associated with inability to break down the
terminal galactose from GM1 ganglioside
Is associated with inability to break down the
terminal galactose from GM1 ganglioside
![]() Results in accumulation of GM1 ganglioside in lysosomes, which are
consequently greatly enlarged.
Results in accumulation of GM1 ganglioside in lysosomes, which are
consequently greatly enlarged.
Enlarged
lysosomes accumulate in the cells of several tissues, the most important of
which are:
![]() Involvement
of
neurons of the central nervous system
Involvement
of
neurons of the central nervous system
-fits, psychomotor deterioration, mental
retardation
![]() Enlargement
of liver & spleen
Enlargement
of liver & spleen
![]() Widening
of bones: deposition in marrow
Widening
of bones: deposition in marrow
![]() Vacuolated
lymphocytes deposition in lymphocytes
Vacuolated
lymphocytes deposition in lymphocytes
This disease is inherited as an autosomal recessive disorder.
Other Lysosomal Storage Diseases
There are several lysosomal storage
disorders. All are associated with a
deficiency of a particular lysosomal enzyme, resulting in accumulation of an
undigested substrate within the lysosomes. The following are a few examples of
lysosomal storage disease with the associated substrate:
![]() Pompe à glycogen
Pompe à glycogen
![]() Hunter
disease à heparan & dermatan sulphates
Hunter
disease à heparan & dermatan sulphates
![]() Morquio’s
disease à keratan& chondroitin sulphate
Morquio’s
disease à keratan& chondroitin sulphate
![]() Tay
Sachs disease à GM2 ganglioside
Tay
Sachs disease à GM2 ganglioside
![]() Niemann-Pick
à sphingomyelin
Niemann-Pick
à sphingomyelin
![]() Farberdisease
à ceremide
Farberdisease
à ceremide
Peroxisomes or Microbodies
The features of peroxisomes are:
 Membrane-bound spherical organelles
Membrane-bound spherical organelles
Have a diameter of 0.2-2.0mm
Contain a granular matrix and an electron-dense crystalline core
Contain the enzymes catalase and urate oxidase
Have the ability to break down H2O2
Have a pH optimum of 7.5
Oxidases produce H2O2
RH2 + O2 à R + H2O2
Catalase decomposes H2O2
- by conversion to water
2H2O2 à H2O
+ O2
- by oxidation of another organic compound
H2O2 + AH2 à 2H2O2 + A
H2O2 is both produced and degraded in peroxisomes.
H2O2 is harmful to cells and has to be degraded almost as fast as it is produced.
Many substances are broken down or metabolised by oxidative reactions in peroxisomes.
The
most important ones are:
1. b-Oxidation of fatty acids
very long are chain fatty acids are broken down to acetyl CoA with the production of H2O2
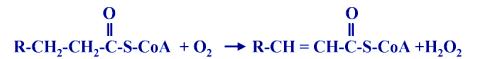
This reaction also occurs in mitochondria.
2. Transfer of NH3 (amino) groups from amino acids to ketoacids
- requires the enzyme aminotransferase
3. Oxidation of uric acid or urates to allantoin
requires the enzyme urate oxidase
All are associated with the production of
H2O2
Peroxisomes
also have synthetic functions including:
- synthesis of cholesterol and dolicholol (also occurs in SER)
- synthesis of plasmalogens (membrane components in brain & heart)
- synthesis of bile acids
Some
diseases are caused by lack of specific peroxisome enzymes:
·
Adrenoleukodystrophy
- a progressive neurological disorder
- a defect
in b-oxidation
of long chain fatty acid,
- results in accumulation of long chain
fatty acids in neurons and other cells
Gout due to accumulation of uric acid
·
- failure of conversion of uric acid to
allantoin
- causes deposition of uric acid in joints,
and consequent painful arthritis
Zellweger syndrome caused by absence of peroxisomes
- causes severe neurological disorder, metabolic defects, and early death
Melanosomes
·
Melanosomes are the organelles in which melanin synthesis occurs.
They are :
- Oval-shaped , membrane-bound bodies
- 0.3-1.3 mm in diameter
- found in melanocytes (pigment-producing cells)
- contain enzymes for the biosynthetic pathway of melanin
- are identified by the DOPA reaction
Biosynthetic pathway of melanin formation
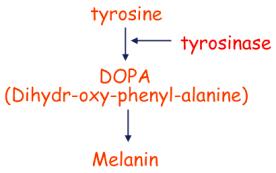
A multi-step pathway in
which tyrosine is DOPA by a series of reactions involving the enzyme tyrosinase
as well other enzymes. DOPA is then
converted to melanin.
In the DOPA
reaction, cells are exposed to an excess of DOPA. Melanin pigment will be
formed in the melanosomes, the organelles that have the necessary enzymes for
the biosynthesis of melanin.
Morphological pathway of melanosome formation
Melanosomes are formed from two sources:
From the SER – pre-melanosomes are formed by coalescence of vesicles, and appear to contain parallel filamentous contents.
In the RER and Golgi complex, tyrosinase and other enzymes are synthesised and packaged into vesicles. These fuse with the pre-melanosomes.
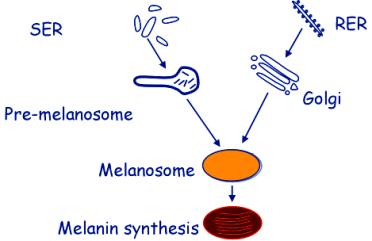
Melanosomes are stimulated by Melanocyte Stimulating Hormone (MSH)
The plasma membrane of melanocyte has receptors for MSH. The MSH enters the cells by receptor-mediated endocytosis and the MSH-containing vesicles fuse with the melanosomes.
Melanin can be transferred from melanocytes to keratinocytes in the skin.
Melanocytes have numerous dendritic processes.
Melanin-containing vesicles are pinched off from the tips of these processes.
They are endocytosed by the keratinocytes, degraded by lysosomes and the melanin become dispersed within these cells.