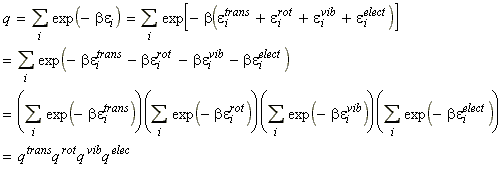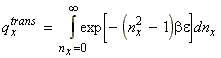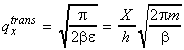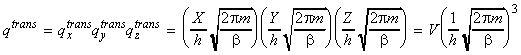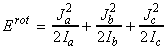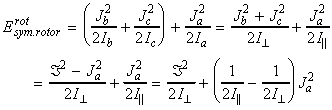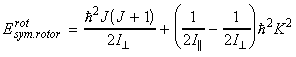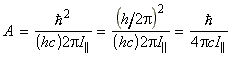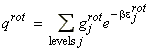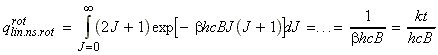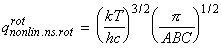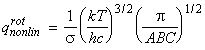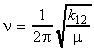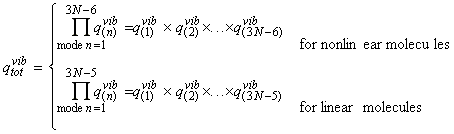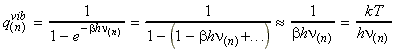|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
Statistical Thermodynamics (ii) The molecular partition function (3-2) The rotational contribution (3-3) The vibrational contribution (3-4) The electronic contribution (3-5) The overall partition function [ TOP ]
(3) The molecular partition function
We may assume that the energy of a molecule is the sum from contributions from the different modes of motion, namely translation, rotation, vibration and electronic contribution, i.e.: or: for short. This means that we are making the assumption that these modes are independent of each other (this is not true, except for translation, but in most cases, this approximation is valid). This means that we may write the partition function as:
[ TOP ]
In classical mechanics, the translational kinetic energy of a particle can assume any value (continuos function). However, at the atomic level, this assumption cannot be made, and instead we have to make use of quantum mechanics, and place limits on the actual kinetic energy that a molecule can have. In particular, if we wish to find what sort of translational energy values a molecule can have, we assume that our molecule is in a box of dimensions X, Y, Z. In such cases, the permitted energy levels ex for translational motion in the x-direction of a molecule of mass m is constrained by two infinite boundary potentials at x = 0 and x = X, and is given by: or relative to the first energy level: where nx is the translational quantum number in the x-direction, and similarly for the y- and z-directions. Thus, since in this case, g(n) = 1, we have:
which gives: Thus, in analogy to above, we have:
which is usually written in the form: where V is the volume, and L is called the thermal wavelength of the molecule is given by: Note: (i) qtrans approaches infinity as T approaches infinity since a large number of states becomes available at very high T. qtransis 0 at 0K. (ii) Typical values qtrans at room temperature: 2 x 1028 for O2 in a 100cc vessel. (iii) Approximations made remain valid if L
>> V. For example, for H2 at 25oc,
L
=
0.71Å, which is comparable to the pores in zeolites (5 Å).
[ TOP ]
As in the case of translational motion, the rotational motion of a molecule
is also quantized. In our discussion of rotational energy, we need to initially
suppose that our molecules are rigid rotors, that is bodies that
do not deform under the stress of rotation. Rigid rotors may be classified
into four types, according to their symmetry/moments of inertia (about
three orthogonal axis):
(i) spherical rotors, (molecules that belong to cubic or icosahedral groups, e.g. CH4, SiH4 and SF6) have three equal moments of inertia 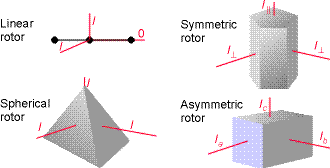 Fig. A schematic illustration of the classification of rigid rotors.
(A) Finding the allowed energies for the different rotors
In classical mechanics, the energy for a rigid body rotating about an axis a is given by: where wa is the angular velocity about the a-axis (units: rad s-1) , and Ia is the moment of inertia about the same axis. Thus for a rigid body free to rotate about the three ortho-axis, we have:or in terms of the angular momentum Ja about the a-axis (Ja = Ia wa), we have:Although in most everyday scenarios Erot and Ja can take any value (classical mechanics / continuum), at the atomic level, this assumption cannot be made, and instead we have to make use of quantum mechanics, and place limits on the actual values of rotational kinetic energy and angular momenta a molecule can have.
(i) The allowed energies for a symmetric motor is dependent o the values of J and K. In particular, for K ¹ 0, each level is doubly degenerate since:
(i) K ¹ 0, the energy levels are 2(2J+1)-fold degenerate; (i) The allowed energies for a symmetric motor (A=B) is dependent on the value of J but not of K However, the quantum number K may still take any of its 2J+1 values. This introduces a (2J+1)-fold degeneracy arising from the orientation in space relative to an arbitrary internal axis.
Given this information of the energy levels and levels of degeneracy, we should now be in a position to compute the partition function, i.e.: For example, let us consider the case of a linear non-symmetric rotor, where:andIn this case, the lower rotational energy level (J=0) is 0, and hence eJ = E. [ TOP ]
(3-3) The vibrational contribution We shall commence our discussion of discussion of vibrational terms by considering a diatomic molecule and model this using the classical ball and spring model, where the two atoms (the balls) have masses m1 and m2 respectively and are connected by a spring of force constant k12. In such case, the classical vibrational frequency (simple harmonic oscillator) is given by: where m is the reduced mass given by: However, as in all other cases, at the atomic level, only quantized frequencies and are allowed, given by:
Polyatomic molecules can undergo may independent vibrational motions. Normal mode analysis (i.e. assuming that an independent, synchronous motion of atoms or groups of atoms within a molecule may be excited without leading to the excitation of any other normal mode) allows us to assume that each normal mode behaves as an independent simple harmonic oscillator. In general, a polyatomic molecule of N atoms has 3N degrees of freedom, three of which are assigned to translation of the molecule as a whole, and another 3 (or 2 if molecule is linear) to rotation. This means that a polyatomic molecule of N atoms has (i) 3N-6 normal modes of vibration if it is non-linear, and (ii) 3N-5 normal modes of vibration if it is linear. Let us now turn our attention to the vibrational partition function. Because we now have 3N-6 (or 3N-5 for linear molecules) normal modes of vibration, our partition function must be a product of the 3N-6 (or 3N-5 for linear molecules) vibrational partition function of the individual modes, i.e.: Let us thus concentrate on a single individual mode term of this partition function, say the one corresponding to the nth mode, with an associated frequency of n(n) Since we are assuming that e0 = 0, then we have e1 = h n(n), e2 = 2hn(n), e3 = 3hn(n), etc. Thus we have: In this case, the energy levels are too far away to approximate by a continuum. However, the summation is very easy since from: we have: where qV is the characteristic vibrational temperature, defined by: where k is Boltzmann's constant. Note that when qV is much larger than the temperature T, then the vibrational partition function will be close to 1, as usually the case (i.e. usually, the molecules are in their ground state w.r.t. rotations. Typically, qV is ~3000K, i.e. qV/T is ~10, which gives qvib = 1/(1-e-10) »1.) This is in sharp contrast with qtrans »1030 and qrot »10. Also, when the bonds are so weak that hn << kT (i.e. |-bhn|<1) or at high temperatures, we may expand the exponential term using the expansion: to get a linear relationship between qvib and T. i.e.: or:
[ TOP ]
(3-4) The electronic contribution Bu the 'electronic contribution' we are referring to the scenario when an electron moves from one orbital to another. However, the separations of the energy levels are usually too big to be overcome simply by thermal excitation. However, there are some exceptions to this statement, namely: (a) In some cases, we have n-fold degenerate ground sates (e.g. the alkali metals, have a 2-fold degenerate ground state, corresponding to two orientation of the electronic spin). In such cases, the partition function is equal to the degree of degeneracy, i.e.: (b) In some cases, there are low-lying electronically excited states. For example, NO, (which has one electron more than N2) we have a scenario where (i) the orbital angular momentum can take two orientation with respect to the molecular axis, and, (ii) for each orientation of the orbital angular momentum, the spin angular momentum may once again take two orientation, hence giving a total of 4 states. The energy of states where the spin and orbital angular momenta are parallel, is slightly greater than the states when they are anti-parallel, a separation arising from spin-orbit coupling, which is only 121cm-1, i.e. at normal temperatures, all four states are thermally accessible. This if we denote e o=0 and e 1=e, then we get:
[ TOP ]
(3-5) The overall partition function Given the four contributions to the partition function, we may now compute the overall molecular partition function as the product of the four contributing partition functions, i.e.: Approximations in our derivation that could lead to errors include: (1) We have assumed that the rotational energy levels are close to each other. (2) We assumed that the vibrational levels are harmonic. Such approximations and errors can be avoided by using energy levels
that are determined spectroscopically, and then evaluate the sums explicitly.
|
| E-mail me at jgri1@um.edu.mt |