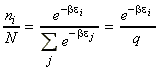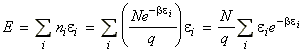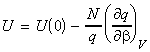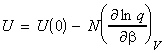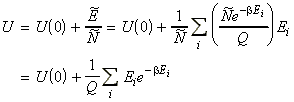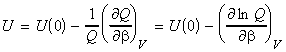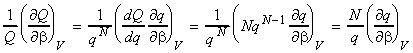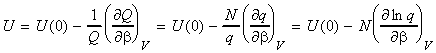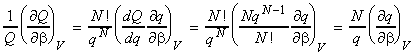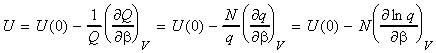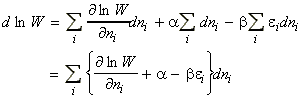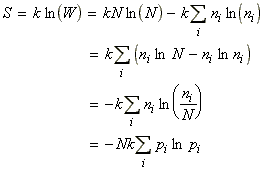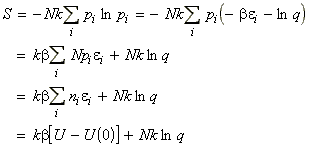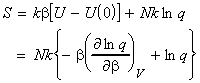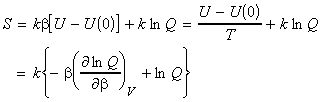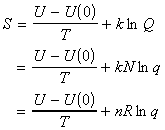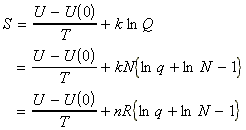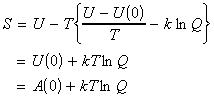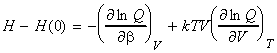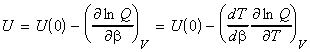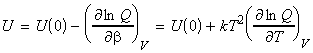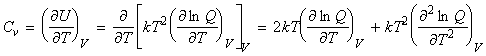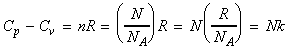|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
Statistical Thermodynamics (ii) (5) Calculation of the various properties from the partition function (5-2) The statistical entropy (5-2-2) Derivation of the statistical entropy in terms of the partition function (5-2-3) Residual entropies (5-4) The pressure (5-5) The enthalpy (5-6) The Gibbs energy (5-7) The heat capacities [ TOP ]
We have said in the discussion above that the partition functions q (valid for independent molecules) and Q (valid for dependent and independent molecules) carry all the thermodynamic information of the system. This is a very strong statement, since the partition functions where derived by looking at the system from the molecular level (statistically) whist the thermodynamic properties refer to the properties of the bulk. Let us illustrate how this relationship between the bulk properties and the molecular level properties q and Q are related with some examples. The derivation of properties in terms of q is more 'basic' and hence, we shall produce full derivations for U and S in terms of the molecular partition function for distinguishable independent particles, q. We shall then proceed by another derivation, a general one in terms of Q, the canonical partition function.
[ TOP ]
(5-1) The internal energy
The total energy of the system relative to the zero point energy* is given by: (*) Recall that we has arbitrarily chose to set the lowest energy level (ground state) at 0J. For distinguishable independent molecules, we have seen before that since the most probable configuration (i.e. the one with a highest W) is so strongly dominating, then we may re-express ni in this energy equation in terms of the Boltzmann distribution, i.e. since (assuming q = qtot): we have: But mathematics tells us that: i.e. assuming that the partition function is only a variable of b (see below), we have
where U(0) is the zero point energy, that is the energy correction that we need to make since in our derivation of q we had assumed that the energy of the lowest states is equal to 0, i.e. e0 = 0. This energy correction equates to the energy at T=0K, since at this point, all the molecules are in their lowest energy states. We should also not that the partition may also depend on other variables apart from T (e.g. volume). In particular, we can replace the full derivative of the partition function by a partial derivative where all the other variables apart from T (or rather b) are kept constant, i.e.: or in terms of ln(q) by: (since dq/q = ln q )
NOTE: A similar relationship may be derived for dependent molecules in terms of the canonical partition function, i.e.: But we know that: whist for most dominant canonical configuration (i.e. the most important one): i.e.: which means that (see derivation using q):
NOTE: An expression of U in terms of the molecular partition function can be obtained from the expression of U in terms of the canonical partition function by recalling that: (1) For independent distinguishable particles, Q = qN , i.e.: i.e.: (2) For indistinguishable particles, Q = qN / N! , i.e.: i.e. as in the case of independent distinguishable particles,
[ TOP ]
(5-2) The statistical entropy
In this section we shall first justify one of the most important entropy relationships in thermodynamics, that is, and then show how S can be derived from the partition functions. We shall then also have a look at the residual entropy, that is, the entropy that is presnt at OK due to 'imperfections' in the crystal structure.
[ TOP ]
From our internal energy equation we had seen that: i.e.:Now since (a) the energy levels do not change when the system is heated at constant volume (they would have changed if work would have been done on the systems), but (b) the populations change, then, in the absence of all changes other than heating:But we know that under the same conditions:i.e.:The most dominant/important configuration (and the only one we need considering) is the one which has maximum W, or maximum ln(W)). This configuration may be obtained by solving:dW=0 or d[ln(W)] = 0where:with the additional constraints that:These three equations may be combined using the Lagrange method of undetermined multipliers to get (using arbitrary constants -b and a to get:
[ TOP ]
(B) Derivation of S from the partition functions.
As in the case of the internal energy U, we shall commence our discussion by deriving the deriving S in terms of q, the molecular partition function for a system of N independent distinguishable particles. [ TOP ]
(C) Residual entropy As we have said in the discussion of entropy in classical thermodynamics, we sometimes make the assumption that the entropy of a system at 0K is zero. However, this is only true for perfect crystals, and in practice, such perfection cannot be achieved. This 'entropy at 0K', S(0) is referred to as the residual entropy. [ TOP ]
(5-3) The Helmholtz free energy NOTE: From now onwards, the derivations shall only be presented in terms of the canonical partition function Q. The Helmholtz free energy A (the free energy at constant volume) is defined as: where A(0) = U(0) and in terms of the canonical partition function, we have: Thus, [ TOP ]
(5-4) The pressure We has seen from classical thermodynamics that: and thus, since A is a state function, we get: which for a reversible path for U (U is a state function, so DU is path independent), we have: (NOTE: In this equation dq and dw refer to heat transfer and work done respectively). Thus, we have: which from the expression of A in terms of the canonical partition function Q we have:
[ TOP ]
(5-5) The enthalpy The enthalpy H of any substance may be derived from: i.e.: [ TOP ]
(5-6) The Gibbs free energy The Gibbs free energy G any substance may be derived from: i.e.:
[ TOP ]
(5-7) The heat capacities The heat capacity at constant volume may be derived from: But where: i.e.: which differentiates to: i.e.: Thus, Cp may then be derived from Cv by recalling that: |
| E-mail me at jgri1@um.edu.mt |