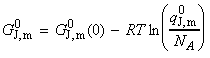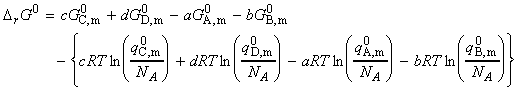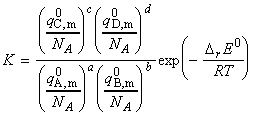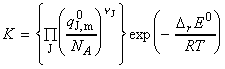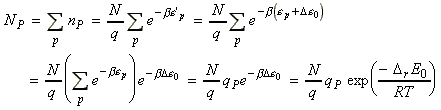|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
| . |
Statistical Thermodynamics (vi) The equilibrium constant (a) Derivation of the equilibrium constant in terms of the partition function [ TOP ]
(a) Derivation of the equilibrium constant in terms of the partition function We have already seen from classical thermodynamics that the equilibrium constant for a reaction is related to the Gibbs free energy of the forward process through: Thus, if we were to consider a gas phase reaction where the equilibrium constant can be expressed in terms of the partial pressures. In this derivation we shall make use of the standard molar Gibbs free energy, defined as: for each of the species J. To calculate this, we need to the value of the standard molar partition function, qm0, that is, the molar partition function when p = p0 = 1 bar. Consider the reaction: The standard Gibbs energy is given by: where: as derived in the previous section (ideal gasses). Thus we have: But G(0) = U(0), the first term on the right equates to the difference in the molar energies of the ground states of the products and reactants and is calculated from the bond dissociation enthalpies (seefig. 1), i.e.: whilst since a ln(x) + b ln(y) - c ln(z) = ln(xa) + ln(yb) - ln(zc) = ln(xayb/zc), the second term is given by: 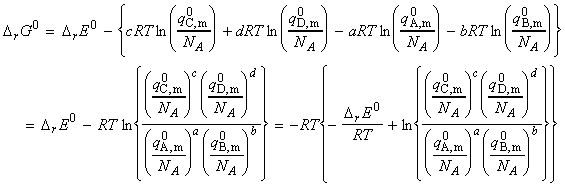
But we know that i.e.: i.e. by taking antilogarithms, we obtain the equilibrium constant: This equation may be summarised by: [ TOP ]  Fig. 1: The definition of DrE0
for the calculation of equilibrium constants.
[ TOP ]
(b) The physical basis for equilibrium constants
In this section we shall look into the physical basis of equilibrium constants, vis-à-vis the simple R <=>P gas-phase equilibrium (R for reactants, P for products). In each of the situations in the fig. 2 below, we have two sets of energy levels: one set of states belongs to R other belongs to P. The populations of the states are given by the Boltzmann distribution and are independent of whether any given state happens to belong to R or to P. We can therefore imagine a single Boltzmann distribution spreading, without distinction over the two sets of states. Then:
The population in a state i of the composite system (i.e. R, P) is given by the Boltzmann distribution, i.e.: where N is the total number of molecules. The total number of R molecules is the sum of these populations taken over the states belonging to R; these states we label r with energies er. The total number of P molecules is the sum over the states belonging toP; these states we label p with energies e`p: where the last step is the conversion of molecular energies to molar energies. The equilibrium constant is hence given by:
Fig. 2: The array of R(eactants) and P(roducts) energy levels:(a)
At equilibrium all are accessible (to differing extents, depending on the
temperature), and the equilibrium composition of the system reflects the
overall Boltzmann distribution of populations. As DrE0
increases, R becomes dominant. (b) It is important to take into
account the densities of states of the molecules. Even though P might lie
well above R in energy (that is, (DrE0
is large and positive), P might have so many states that its total population
dominates in the mixture. In classical thermodynamic terms, we have to
take entropies into account as well as enthalpies when considering equilibria.
NOTE: The significance of this last equation can be seen most clearly by exaggerating the molecular features that contribute to it (see Fig. 3). We shall suppose that R has only a single accessible level, which implies that qR = 1. We also suppose that P has a large number of evenly, closely spaced levels. The partition function of P is then qP = kT/e. In this model system, the equilibrium constant is:

[ TOP ]
|
| E-mail me at jgri1@um.edu.mt |