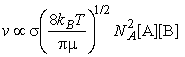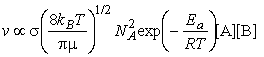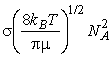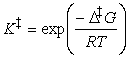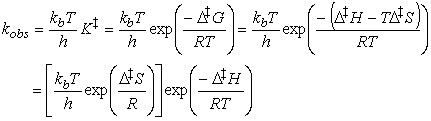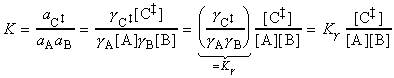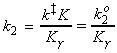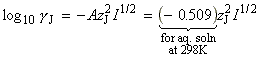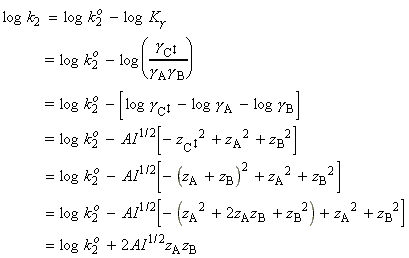|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
A Theoretical approach to reaction rates: The Collision theory and the Activated Complex Theory (2) Activated complex theory (2-1) The reaction profile in the ACT (2-3) The activated complex theory and reactions between ions.
[ TOP ]
(1) Collision theory
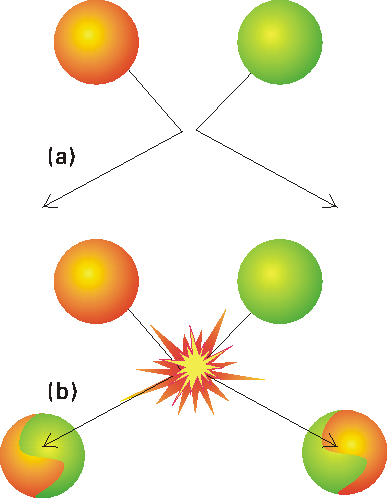
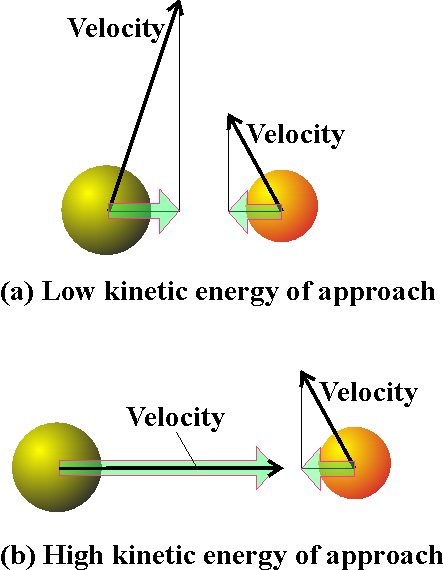
In this section we shall attempt to understand the origin of the Arrhenius parameters by studying a class of gas-phase reactions in which reaction occurs when two molecules meet (collision theory). Under this theory, reaction occurs only if two molecules
collide with a certain minimum kinetic energy along their line of approach
(Fig 1). In fact, in the collision theory,
a reaction resembles the collision of two defective billiard balls:
the balls bounce apart if they collide with only a small energy, but might
smash each other into fragments (products) if they collide with more than
a certain minimum kinetic energy. This model of a reaction is a reasonable
first approximation to the types of process that take place in planetary
atmospheres and govern their compositions and temperature profiles.
Fig. 1: In the collision theory of gas-phase chemical reactions, reaction occurs when two molecules collide, but only if the collision is sufficiently vigorous, (a) An insufficiently vigorous collision: the reactant molecules collide but bounce apart unchanged, (b) A sufficiently vigorous collision results in a reaction. NOTE: The importance of the collision theory is that it provides a proof from first principles that reactions (or, strictly speaking, gas phase reactions!) can indeed display Arrhenius-like behaviour.
[ TOP ]
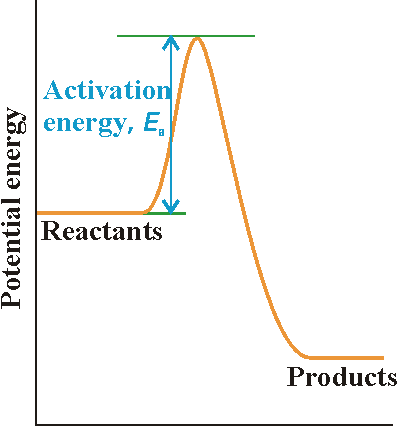
(1-1) The reaction profile in the Collision theory:
A reaction profile in collision theory is a graph
showing the variation in potential energy as one reactant molecule approaches
another and the products then separate (Fig
2). (NOTE: Recall that the potential energy of an object is the
energy arising from its position (not speed), in this case the separation
of the two reactant molecules as they approach, react, and then separate
as products.)
This profile shows:
For the reaction to be successful, the reactant
molecules must approach with sufficient kinetic energy along their line
of approach to carry them over the activation barrier, the peak
in the reaction profile. As we shall see, we can identify the height of
the activation barrier with the activation energy of the reaction.
[ TOP ]
(1-2)
Derivation of the rate law through the Collision theory:
With the reaction profile in mind, we can derive the rate law for a reaction: where:
as follows:
(a)
The encounter rate:
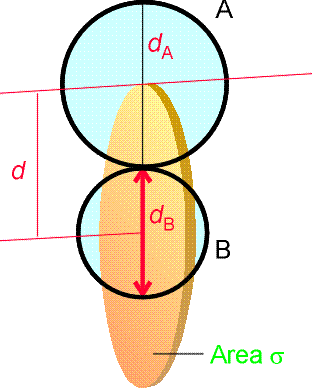
where NA is Avogadros constant, kB is Boltzmanns constant, T is the temperature, s is the collision cross-section (see fig. 3), and m is the reduced mass given by:
(b) The minimum energy requirement:
i.e. at this stage we can conclude that the rate of reaction, which is proportional to the rate of collision multiplied by the fraction of successful collisions, is: Note that this equation now has the Arrehenius form, i.e.: This means that at this stage we can already conclude that The activation energy, Ea, is the minimum kinetic energy required for a collision to result in reaction. (c) The steric factor
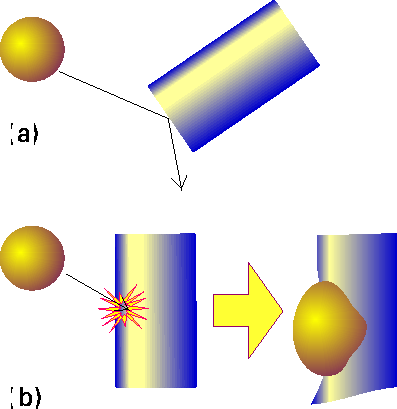
One possible explanation is that, not only must the molecules collide with sufficient kinetic energy, but they must also come together in a specific relative orientation (Fig 5).
E.g. 2: For the hydrogen addition reaction in which a hydrogen molecule attaches directly to an ethene molecule to form an ethane molecule: This very low value of P which suggests that the reaction has very stringent orientational requirements.
In this reaction, the distance of approach at which reaction can occur seems to be considerably larger than the distance needed for deflection of the path of the approaching molecules in a non-reactive collision! To explain this surprising conclusion, it has been proposed that the reaction proceeds by a harpoon mechanism.
The
activated
complex theory (ACT), formerly known as the transition state theory,
is a more sophisticated theory of reaction rates that offers the following
advantages over the collision theory:
NOTES:
(1) The concepts used in the derivation of the rate constants in terms of the ACT are based on statistical thermodynamics. (2) The reason why P appears automatically in the ACT framework is that the orientation requirements are carried in the entropy of activation. Thus, if there are strict orientation requirements (for example, in the approach of a substrate molecule to an enzyme), then the entropy of activation will be strongly negative (representing a decrease in disorder when the activated complex forms), and the pre-exponential factor will be small. In practice, it is occasionally possible to estimate the sign and magnitude of the entropy of activation and hence to estimate the rate constant. (3) The general importance of activated complex theory is that it shows that even a complex series of events (not only a collisional encounter in the gas phase) displays Arrhenius-like behaviour, and that the concept of activation energy is applicable.
[ TOP ]
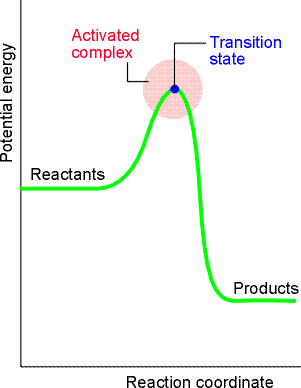
(2-1) The reaction profile in the ACT:
Activated complex theory is an attempt to identify the principal features
governing the magnitude of a rate constant in terms of a model of the events
that take place during the reaction. Let us have a look at a typical the
reaction
profile (i.e.a plot of the 'potential energy' vs. 'reaction coordinate'),
for a bimolecular reaction between A and B forming products P (fig. 6).
This plot illustartes the following features:
Fig. 6: A reaction profile. The horizontal axis is the reaction coordinate, and the vertical axis is potential energy. The activated complex is the region near the potential maximum, and the transition state corresponds to the maximum itself. In other words we have: which in terms for the ACT, we have: or: i.e. the reaction between A and B proceeding through the formation of an activated complex, C, that falls apart by unimolecular decay into products, P. Note that we not all (AB) will proceed to products, and in fact, some will decay back to A and B, i.e. The scope now becomes to derive an expression for k2 from first principles, which as we have said before, is done through concepts from statistical thermodynamics. This formulation is known as the Eyring equation.
[ TOP ]
(2-2)
Derivation of the rate law through the ACT (the thermodynamic derivation):
As indicated above, in a simple form of activated complex theory, we suppose that the activated complex is in equilibrium with the reactants (pre-equilibrium), and that we can express its abundance in the reaction mixture in terms of an equilibrium constant, K. Then,
if we suppose that the rate at which products are formed is proportional
to the concentration of the activated complex, we can write:
The full activated
complex theory gives an estimate of the constant of proportionality to
give:
Thus, if we compare
this expression with the form of the rate law, i.e.:
we would find:
But since we
may think of K as an equilibrium constant, then it may
be expressed in terms of the standard reaction Gibbs energy, or in this
context, the activation Gibbs energy, and written DG.
It follows that:
i.e.:
But:
i.e.:
This expression has the form of the Arrhenius expression,
i.e.:
if we identify the enthalpy of activation, DH,
with the activation energy and the term in square brackets, which depends
on the entropy of activation, DS,
with the pre-exponential factor.
[ TOP ]
(2-3) The activated complex theory and reactions between ions. The simplified (thromodynamic) approach to the activated complex theory simplifies the discussion of reactions in solution. We can combine the rate law: with the thermodynamic equilibrium constant: i.e.: Let us now define ko2 as the rate constant with the activity coefficients equal to 1, i.e.: At low concentrations the activity coefficients can be expressed in terms of the ionic strength, I, of the solution by using the Debye-Huckel limiting law (see P.W. Atkins, Physical Chemistry, 6th Ed. Section 10.2c, particularly eqn 10.19) in the form: where A = 0.509 in aqueous solution at 298 K and zJ is the charge numbers on particle J. Thus,
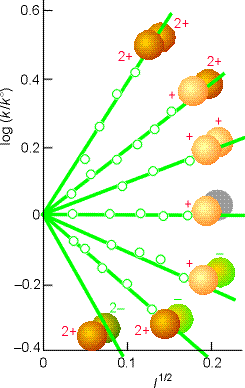
Conversely, if reactant ions have opposite same signs, the charges in the activated complex cancel out and the complex has a less favourable interaction with its atmosphere than the separated ions with a solution of a high ionic strength. >> TECHNICAL NOTE: A plot of log(k/k0)
vs. l1/2 should be linear wheer the gradient gtives infomation
about the charge type of the activated complex for the rate determining
step. (see Fig 5 below).
Fig. 6: Experimental tests of the kinetic salt effect for reactions in water at 298 K. The ion types are shown as spheres, and the slopes of the lines are those given by the Debye-Hückel limiting law and the Kinetic salt effect equation above (Eqn. K.Salt.Effect). |
| E-mail me at jgri1@um.edu.mt |