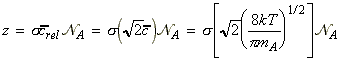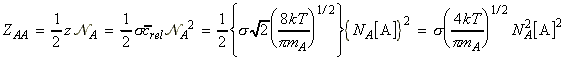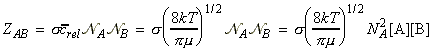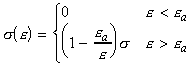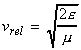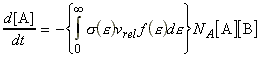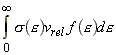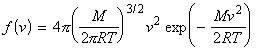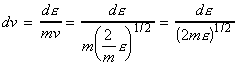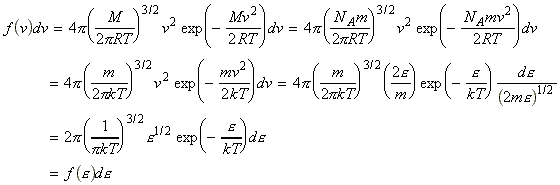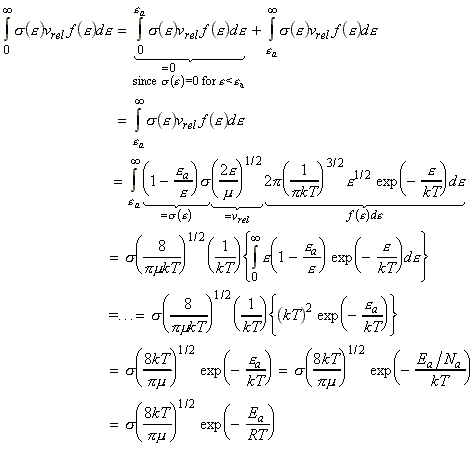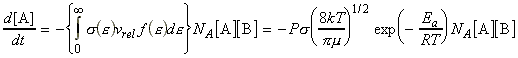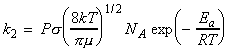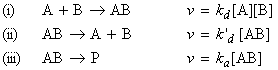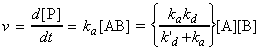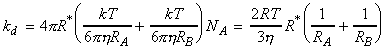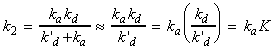|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
CH237 - Chemical Thermodynamics
and Kinetics
Dr.
Joseph N. Grima, Department
of Chemistry
|
|
A theoretical investigation of chemical kinetics - Derivation of the rate constant from first principles (ii) (1) Reactive encounters:
(0) Aside- The kinetic Theory (on a separate page) (1-2) Reactive encounters in the Liquid Phase
[ TOP ]
In this section we shall attempt to derive from first principles an expression for k2, the rate constant for the gas-phase bimolecular elementary reaction below: We can anticipate that the factors that we should consider are:

[ TOP ]
(1-1-1) The encounter rate.
In this section we shall derive an expression for the collision density, ZAB which is defined as:
This is of course related to the collision frequency. We have already seen that the collision frequency, z, for a single A molecule of mass mA in a gas of other A molecules is given by: where s is the collision cross-section, and 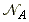 is the number density of A, given by:
is the number density of A, given by:
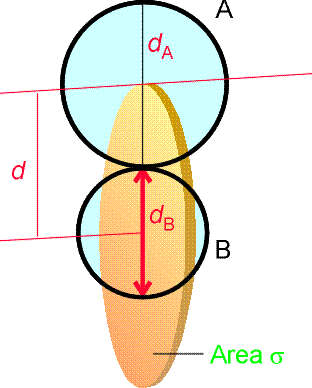
NA being Avogadro's number. Given the collision frequency, z, we may calculate the collision density, ZAA, that is the density of collisions between two A molecules in this gas. This is given by: where the factor 1/2 is introduced to ensure that a collision is not counted twice. (When a A1 collides with A2, then inevitably, A2 collides with A1. However, this is only once collision.) In analogy to all this, the collision density for an A with B collision is given by: (The 1/2 factor has now been discarded because we are considering an A molecule colliding with any B molecule.) In this case, the collision cross-section s is given by (see fig 1.): and m is the reduced mass. Fig. 1: The collision cross-section for two molecules can be regarded to be the area within which the centre of the projectile molecule (A) must enter around the target molecule (B) in order for a collision to occur. If the diameters of the two molecules are dA and dB, the radius of the target area is d = 1/2 (dA + dB) and the cross-section is pd2.
[ TOP ]
(1-1-2) The minimum energy requirement
We can include the minimum energy requirement condition by expressing the collision cross-section as a function of e, the KE of approach (relative KE) of the two colliding species before collisions. We shall set this cross-section s(e) as: This function indicates that if we do not have the energy ea (the threshold value of energy) then s(e)=0. Otherwise, the cross-section will have a value that depends on s and on ea/e. Thus for a collisions with a specific relative speed of approach vrel we have: where the relative KE, e, is related to the relative speed through: i.e.: However since a wide range of approach energies are present in the sample, we should average this expression, as in the case of the Maxwell-Boltzman distribution, i.e.: (Note that in contrast with ZAB , in this expression there is only one Avogadro's constant since we are now dealing with moles, i.e. we have to divide by NA .) Thus the problem reduces to evaluation of the integral: Let us start by finding an expression for f(e)de. From the Maxwell-Boltzmann distribution for molecular speeds: and from k = R / NA and e = ½mv2 i.e. de = mvdv, i.e.: we note that: i.e.: where Ea is the activation energy.
[ TOP ]
(1-1-3) The steric factor
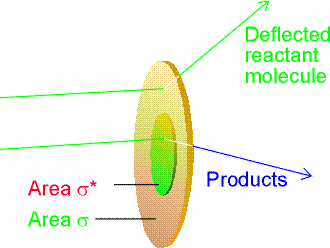
At this point we introduce a steric factor, P, and define the reactive cross-section s* as a multiple of the normal cross-section, (see fig. 2) i.e.: (P is usually (though not always) several orders of magnitude smaller than 1, and in some cases, may be obtained from experiment.) Fig. 2: The collision cross-section is the target area that results in simple deflection of the projectile molecule; the reaction cross-section is the corresponding area for chemical change to occur on collision.
[ TOP ]
Given all these expressions, we may re-write the rate equation as: But we know that: Thus we note that: Note that this equation is in the form of the Arrhenius equation provided that the exponential temperature dependence dominates the square root temperature dependence of the pre-exponential factor. It follows that the activation energy, Ea can be associated with the minimum KE along the line of approach that is needed for a reaction, and that the pre-exponential factor is a measure at which collisions occur in a gas.
[ TOP ]
(1-2) Reactive encounters in the Liquid Phase Encounters between reactants in solution occur in a very different manner from encounters in gases:
Consider the reactions: where AB signifies the 'encounter pair'. This reaction involves three 'steps': (i) the formation of the encounter pair AB form A and B;Thus we have: Using the steady state approximation we have: i.e. assuming equality: i.e.: i.e. The rate of formation of P is given by: i.e.:
(1-2-1) A diffusion-controlled reaction, when ka >> k'd [ TOP ]
(1-2-1) A diffusion-controlled reaction, when ka >> k'd, that is once AB has been formed, then the rate of re-separation of the unreacted encounter pair is much slower than that for the formation of the products. In such case we have: In this diffusion-controlled limit, the rate of reaction is governed by the rate at which the reactant molecules diffuse through the solvent. An indication that a reaction is diffusion-controlled is that its rate constant is of the order of 109 Lmol-1s-1 or greater. Radical and atom recombination reactions are often diffusion-controlled since the combination of radicals involves very little activation energy. This rate of reaction is calculated by considering the rate at which the reactants diffuse together. In particular, if we assume that a reaction will only occur if the two reactant molecules come within a distance R* from each other, then we have: i.e.: where DA and DB are the diffusion coefficients of the two reactants in solution, which can be estimated by the Stokes-Einstein equation as: where h is the viscosity, i.e. (since k = R/NA): If we further assume that In this approximation, the rate constant is independent of the actual reactants and depends only on the temperature and the solvent viscosity. It shows that the rate constant increases with increase in temperature and decreases with an increase in solvent viscosity (reactants find it more difficult to move through solvent).
[ TOP ]
where K is the equilibrium constant for A + B = AB. In this case, the reaction proceeds at the rate at which energy accumulates in the encounter pair from the surrounding solvent. |
| E-mail me at jgri1@um.edu.mt |